This article was originally published as the introduction of my PhD thesis. After receiving favourable feedback to the work, I’m publishing it here after some adaptations to be less thesis specific.
If you want to read my full thesis, you can download it here!
Abstract
Causal relationships provide valuable insights into how individuals work on the inside. In the last decade, we have seen immense progress in the development of large observational cohort studies of many individuals that measure nearly everything, from DNA to molecular measurements, clinical phenotypes, through to eating habits. These studies have provided us with a wealth of information on the correlations between the measured factors. Unfortunately, observational studies have not been able to identify causal relationships because correlations are often distorted by confounding. This thesis attempts to identify causal relationships from observational studies through the use of instrumental variable methods like Mendelian randomization (MR). In this introduction, I provide background on the state of the art of human molecular measurements. I then discuss how observational studies of these measurements are often confounded, and how MR is able to remove this confounding. I explain the theory behind MR, show how it works, and discuss how it fails. Then I will outline all the steps that we have taken to identify causal relationships across a wide range of (molecular) human phenotypes.
Introduction
Broadly speaking, we understand all the basics of human biology. For example, we know that any biological cell is made up of a combination of water, salts, small molecules, proteins, lipids, glycans, nucleotides and trace elements [1]. We also understand how these components are metabolized and synthesized, how cells multiply through mitosis and how a new organism is created through embryogenesis. A newly formed organism divides into multiple cells, differentiates and eventually turns into a full grown adult human. These broad strokes are the molecular basis of our lives, but many details are still lacking.
Researchers around the globe study different aspects of the human organism. And although progress has been made, there are still many aspects and processes we don’t understand. For instance, it is still largely unknown how the most commonly used painkiller, paracetamol, reduces fever and pain [2]. Fortunately, we don’t need a full understanding of what the active compound in paracetamol does in the human body to benefit from its use(1). We also don’t know the causes of most human diseases, in particular the more chronic diseases for which the burden is enormous on society, the health care system and our economy[3].
Understanding the causes of disease allows us to more accurately treat it. Take the recently approved COVID-19 vaccines as an example [4,5]. These vaccines have been designed, developed and validated incredibly quickly. In part because researchers were aware of the infection mechanisms discovered from other corona virus epidemics such as SARS and MERS [6]. Without an accurate understanding of the mechanisms of coronavirus viral infection, it’s unlikely that we would have such effective COVID-19 vaccines so soon after the discovery of the virus [7].
It may be worthwhile now to consider what has brought us to the stage where we know a fair amount about the inner workings of the human cell, and what steps still need to be taken to help us find causal relationships that are important to understand the human organism.
In the last decades, very large cohorts of many different individuals have been set up. These cohorts have been instrumental in identifying risk factors for disease. For instance, a relationship between cholesterol levels and heart disease was initially identified in the Framingham heart study [8]. Nowadays, we have combined these cohorts with an immense amount of phenotypic measurements. Measurements which help us identify which phenotypes may be important for the development of human disease [9–11].
In parallel, there has been enormous progress into measuring molecular phenotypes in high throughput, which allows us to measure genetics, DNA methylation, mRNA expression, proteins and even our microbes with which we have a synergistic relationship. I call these factors “-omics” layers as they measure the genome, epigenome, transcriptome and microbiome of an individual. These -omics layers can also be measured in a cohort specific manner, allowing for the identification of molecular biomarkers through correlation analyses. This has been very successful, and many correlations between different human traits have been found. These correlations are very interesting, but it can be difficult to discern a cause from consequence based on correlations alone. For instance, there have been many observational associations between the molecular measurements of polyunsaturated fatty acids. For instance, Omega-3 fatty acids are correlated to health related outcomes like cancer and cardiovascular disease, however, after trials have been performed, the health effects of Omega-3 intake seem negligible or non-existent [12,13].
In this introduction I will discuss a state of the art for molecular mechanisms, and how they are used to identify risk factors for human disease. Then I will discuss why we cannot rely on correlations between these risk factors to identify causal relationships, and finally, I will discuss how we can use a technique called Mendelian randomization to identify causal relationships from observational traits. After this introduction to the field, I will state the aim of my PhD and outline how I have tried to achieve this aim through the chapters of the rest of this book.
New molecular measurement techniques are opening Pandora's box of human biology
High-throughput molecular measurements using -omics techniques
The availability of more and better ways to measure the human cells has aided in our knowledge of the inner workings of ourselves. If we consider how much work has been done to determine the basic building blocks of human physiology, we are standing on the shoulders of giants. Presently, we can measure most cellular molecules relatively inexpensively with high throughput. For instance, the most common genetic variations(2) on the human genome are easily measurable using genotyping arrays for tens of euros per sample [14]. For a little more money (a few hundred to a thousand euros), it is possible to measure the full genome as well as all of an individual’s mRNA gene expression(3) (transcriptome) and epigenetic marks (epigenome) through sequencing techniques [15–17]. On top of that, we are in the midst of a single cell revolution, where we are able to identify the genetics, expression and epigenetics of individual cells [18,19]. All this high-throughput sequencing can be applied to any organism, allowing us to also characterize the microbiome, the community of bacteria, virus and fungi and their metabolites that inhabit body sites, such as gut or skin, all of which could be involved in disease onset and progression [20].
The availability of all these layers of information has opened up the study of so called -omics data(4). Nowadays, most large population studies or cohorts at least include DNA measurements of their participants next to a variety of directly measured phenotypes and deep metadata information recorded through questionnaires [9,10]. Many cohorts go even further, developing so-called multi-omics(5) cohorts, in which multiple data layers are measured, from the genome through mRNA expression to the microbiome [11,21,22].
In this time of high-throughput measurements of tens of thousands of molecular phenotypes across thousands to hundreds of thousands of individuals, it would seem that we’re in a perfect position to understand everything there is to know about human biology.
Genome wide association studies map phenotype variation to locations on the DNA
Unfortunately, measuring all molecules in humans will not directly help us in understanding how human disease occurs. Just measuring a molecule will not be enough, these molecules need to be put into the context of other human traits.
In the last two decades, one popular approach was to start with an easily measurable (quantitative) or clinically relevant phenotype, such as cholesterol level, or a clinical outcome like a disease, and identify which markers on the DNA are likely to be involved in the modulation of that quantitative phenotype or the development of the disease. For example, if we’re interested in a disease like celiac disease(6), we could gather a large group of individuals diagnosed with celiac disease, and a large control group who have not been diagnosed with celiac disease, and measure their DNA (Figure 1). We can then compare the prevalence or frequency of each variant on the DNA between the group of cases and the group of controls. If some alleles are much more prevalent in the disease group compared to the control group, we could conclude that this variant is implicated in disease onset. Technically speaking, we say that this variant is associated with the disease (Figure 1) [23] .
This method is called a genome-wide-association study (GWAS), and hundreds of diseases or complex phenotypes have been studied with this approach [24]. The locations on the DNA that are associated with the disease are very informative, because we know which genes and regulatory elements reside in these DNA locations, and they have been associated with our disease of interest.
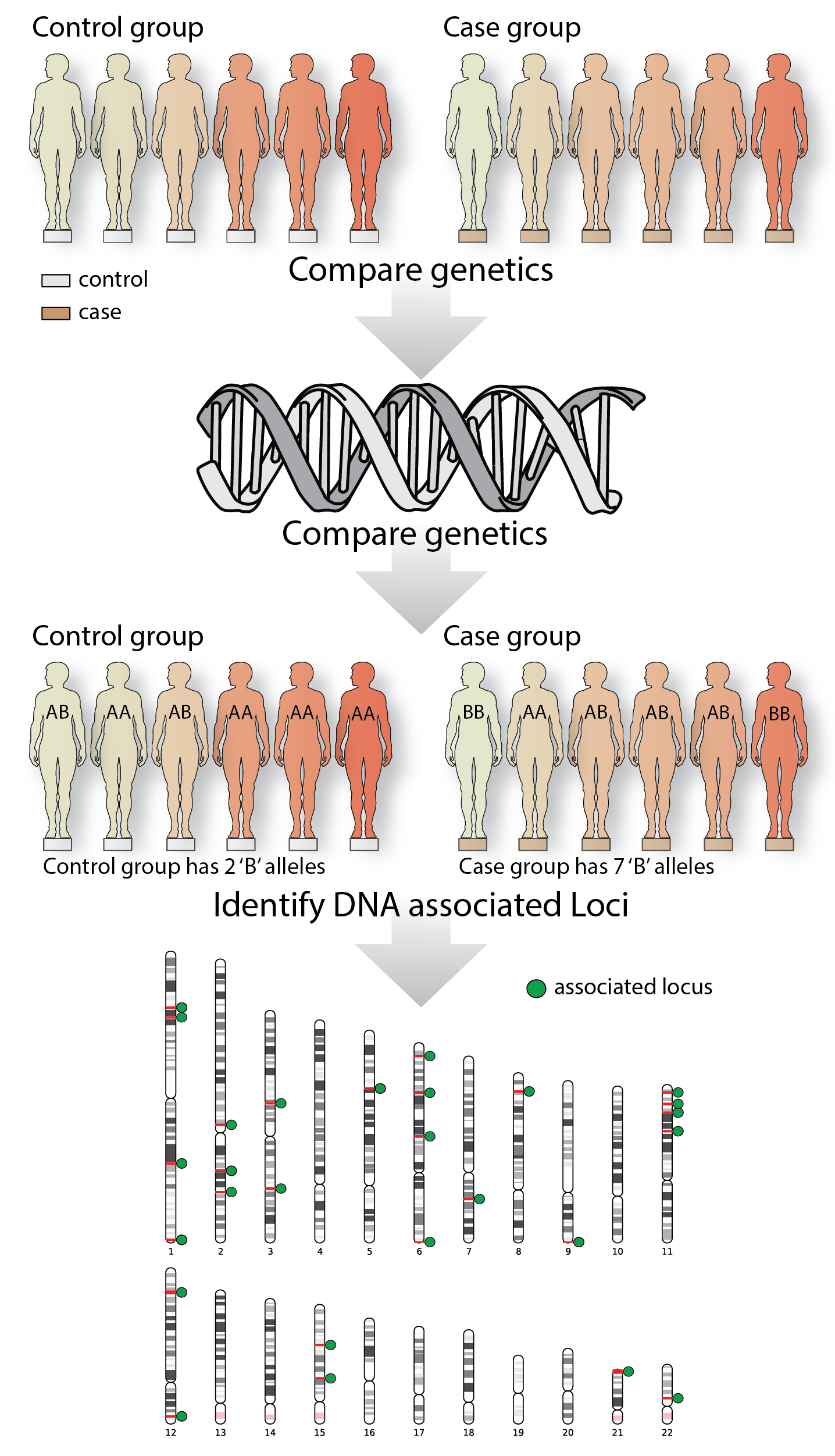
Figure 1: A simple GWAS study
Genome-wide association studies determine the genetic locations on the DNA that are associated with a trait. In this fictional example, a case and control group are recruited, their DNA is measured, and the allele frequencies between the two groups are compared. For ease of the argument, these individuals only have one allele, whereas in the real world, individuals are diploid.
Interpreting GWAS can be difficult
GWAS studies have been able to identify thousands of locations on the DNA that are associated with human phenotypes (GWAS locus or quantitative trait loci (QTL)). In most of these loci, often many (tens to hundreds) genetic variants are associated with the phenotypes with strong statistical evidence and identifying the mechanism that causes the association is difficult. In some cases, explaining the causal role of a DNA location is trivial: if the most-associated variant is a protein-changing sequence, this leads to the strong claim that this is the causal variant and the disease or the quantitative trait is caused by a specific protein isoform(7). Interestingly, however, most GWAS loci are located in non-coding locations of the genome [23,25,26], meaning that they do not alter the final sequence of any proteins. Therefore, figuring out which (molecular) biological factor is causal to the disease requires further analyses or experiments. In the last decade there has been a strong push to try to understand the causal variants and genes that GWAS loci arise from, and how they transfer onto the final phenotype of interest.
Several approaches exist to interpret GWAS loci. One popular class of methods is gene-set analysis [27,28], whereby the genes or other DNA features that are close to the GWAS hits are analyzed in an enrichment analysis. These aggregate analysis techniques are very suitable for describing biological function but will not be able to pin-point causal mechanisms at a single locus. To really describe the function of a single GWAS locus. We have to move to other techniques.
GWAS studies can be combined to interpret GWAS loci
Luckily we can use GWAS to understand GWAS. An interesting feature of a GWAS analysis is that it is largely agnostic to the phenotype of interest. There is only a single requirement: the phenotype of interest needs to be heritable(8). Given this minimal requirement, GWAS have been done on almost everything, from human height, to questionnaire data, to psychic ability(9) [29,30].
If GWAS results are available for two traits, we can compare the associated GWAS loci to see if they share the same causal genetic variants. This is called GWAS colocalization. Multiple methods exist to perform GWAS colocalization, and they have been widely, and successfully used to identify which variants overlap on the genome [31,32].
Colocalization methods are particularly helpful when applied to -omics molecular measurements in conjunction with a quantitative phenotype or disease. We expect that some molecules are an intermediate causal step towards the disease (Figure 2A), which also means that the molecular trait should share the same causal variants with the disease. In this way colocalization methods can estimate causal relationships, although it is difficult to claim that one trait is causal for the other based solely on the sharing of a causal variant. For instance, both traits could share causal variants by chance (Figure 2B).
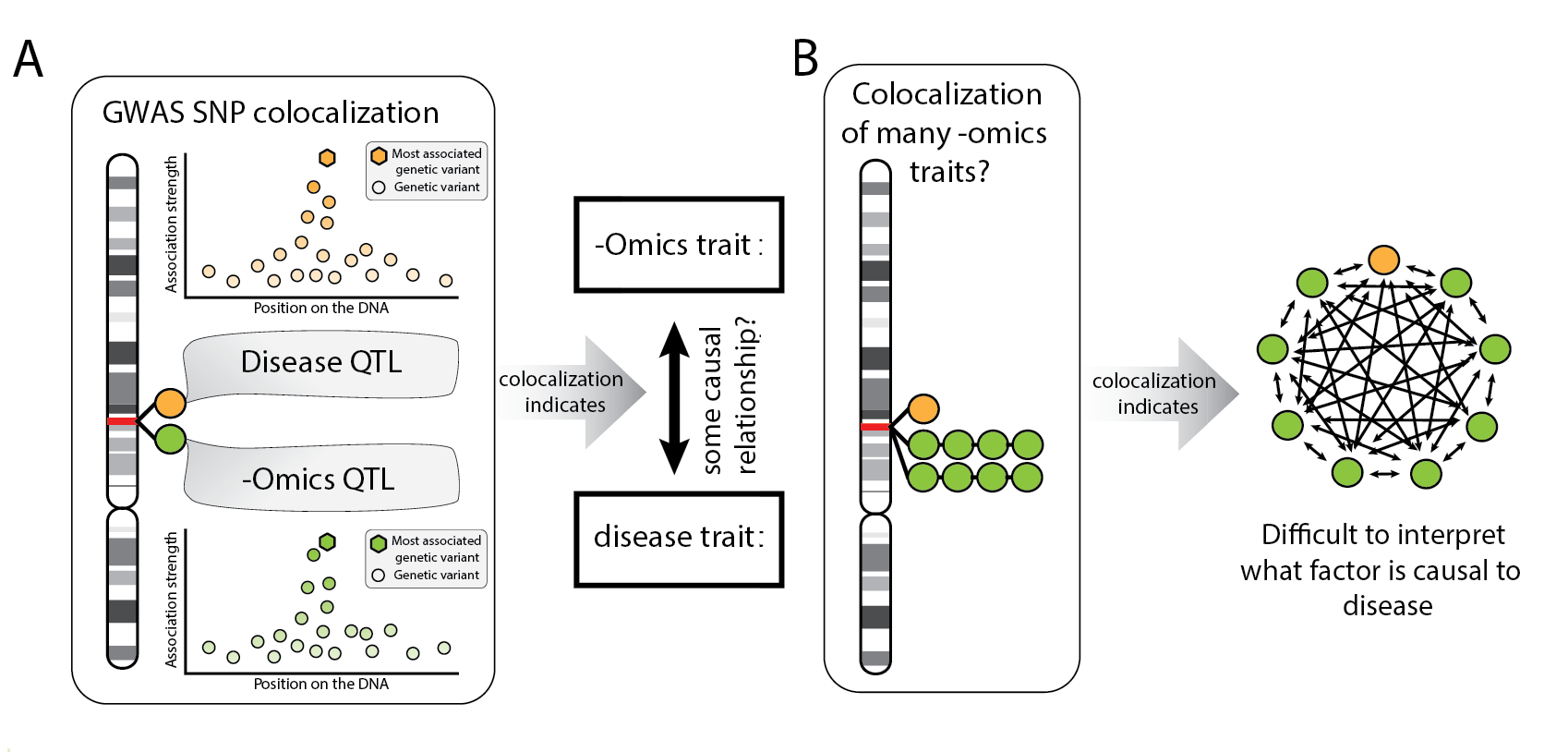
Figure 2: Explanation of colocalization methods
(A)Colocalization methods can be useful to identify causal hypothesis. (B) If many phenotypes colocalize in the same locus, it’s difficult to interpret what the causal relationships could be. Any direction is, in principle, possible.
Interpreting GWAS remains difficult
Protein-coding variants, Gene set analysis and colocalization approaches have been widely applied to analyze GWAS results. These analyses have been able to derive interesting hypotheses on the causal elements of some GWAS loci, but the majority of the causal mechanisms underlying these loci remain unexplained even after using these techniques.
It is striking that we have been so successful in figuring out which genetic locations are important for human traits, but have not been able to discern how these loci work. One potential explanation for why some GWAS loci remain unexplained is that we’re not measuring the correct molecular markers, or we’re not measuring them in the correct context. For instance, when looking at lipid traits, the relevant tissue is more likely to be the liver than blood, because lipid metabolism mostly takes place in the liver [33]. This can be an issue in difficult-to-access tissues like brain, where a biopsy can only be done post-mortem. Next to tissue accessibility, there is also the issue of when measurements are taken. With immune responses, tissues need to be stimulated before the immune response can be seen. Similarly, with phenotypes that develop and mature early, like human standing height, if we measure a molecular layer when growth has stopped, the dynamics of the phenotype are not taken into account in the analysis.
Another reason why GWAS loci remain unexplained is simply the sheer number of loci that have been found. Almost all parts of the human genome are mapped to some trait or another. Single (non-protein-coding) genetic variants can be associated with tens or even hundreds of different other phenotypes [34,35]. Clearly, applying colocalization methods on these hits will naturally produce results that are very difficult to interpret, because there may be evidence for a shared causal variant associated with too many phenotypes.
This is not to argue that GWAS has been an exercise in futility. GWAS analyses have led to profound insights into many different diseases, and have shown how phenotypically different traits relate to each other genetically and, by extension biologically. Furthermore, GWAS have paved the way toward -omics and high-throughput analyses of molecular systems. Still, GWAS remains an observational study, that is not designed to identify causal relationships on it’s own. Unfortunately, the techniques mentioned in this section cannot pinpoint causal relationships with certainty.
Causal relationships from experiments and observational studies
Experimentation is required to understand mechanisms in human biology
Strictly speaking, a GWAS cannot find a causal variant. Nor will colocalizing the GWAS loci with a molecular trait assure that we will find a true biological causal mechanism, given the issues described above. It is therefore crucial to implement systematic approaches that could be used to robustly identify causal relations and inspect several hypotheses in a limited amount of time.
Our understanding of (human) biology is due mostly to experiments and long term epidemiological studies [36,37]. In these experiments, researchers aim to identify a causal relation between a factor and an outcome by comparing one group that has been exposed to the factor with a control group. In this scenario, the null hypothesis would state: if there is no causal relationship, there should be no difference in the outcome between the two groups after a certain time. When this is not true, the null hypothesis is rejected, and there seems to be a difference between groups that is due to the factor to which one of the groups has been exposed (Figure 3). An example of such an experiment is a study interested in the effect of a new drug proposed to reduce the chance of heart disease. The drug can be said to work if the untreated group has a higher incidence of heart disease compared to the treated group (Figure 3).
We call these types of experiments ‘treatment‒control’ experiments, and they are designed for the identification of so-called causal relationships (Figure 3). Causal relationships are extremely informative because they reveal how one factor affects another, for instance the effect of a new cholesterol-lowering medication on the incidence of heart disease.
These treatment-control experiments don’t find all the causes that define a biological trait, rather they identify contributory causes. Evidence found in experiments does not provide proof that the new drug will always work against the disease [38]. An experiment can only show that the chance of heart disease onset is smaller when the drug is taken, compared to when the drug is not taken. Likewise, contributory causes do not exclude other factors from also causing a reduction or increasing the incidence of heart disease. You remain at higher risk for heart disease if you are, for instance, an avid smoker.
Causal relationships are critical for discovering potential treatments. If a relationship is causal, say LDL-cholesterol levels are causal for heart disease, the perturbation of LDL cholesterol will also have an effect on heart disease incidence. For this reason, causal relationships are important in the identification of potential therapeutic targets, and thus for improving improving patient care and disease prevention. A causal relationship also automatically shows how one factor influences another, providing essential information for the identification of biological mechanisms and pathways (38). Pathway information can be used to understand human biology, but, perhaps more interestingly, broad integration of all known pathways will provide further understanding into disease courses and other therapeutic approaches for other diseases.
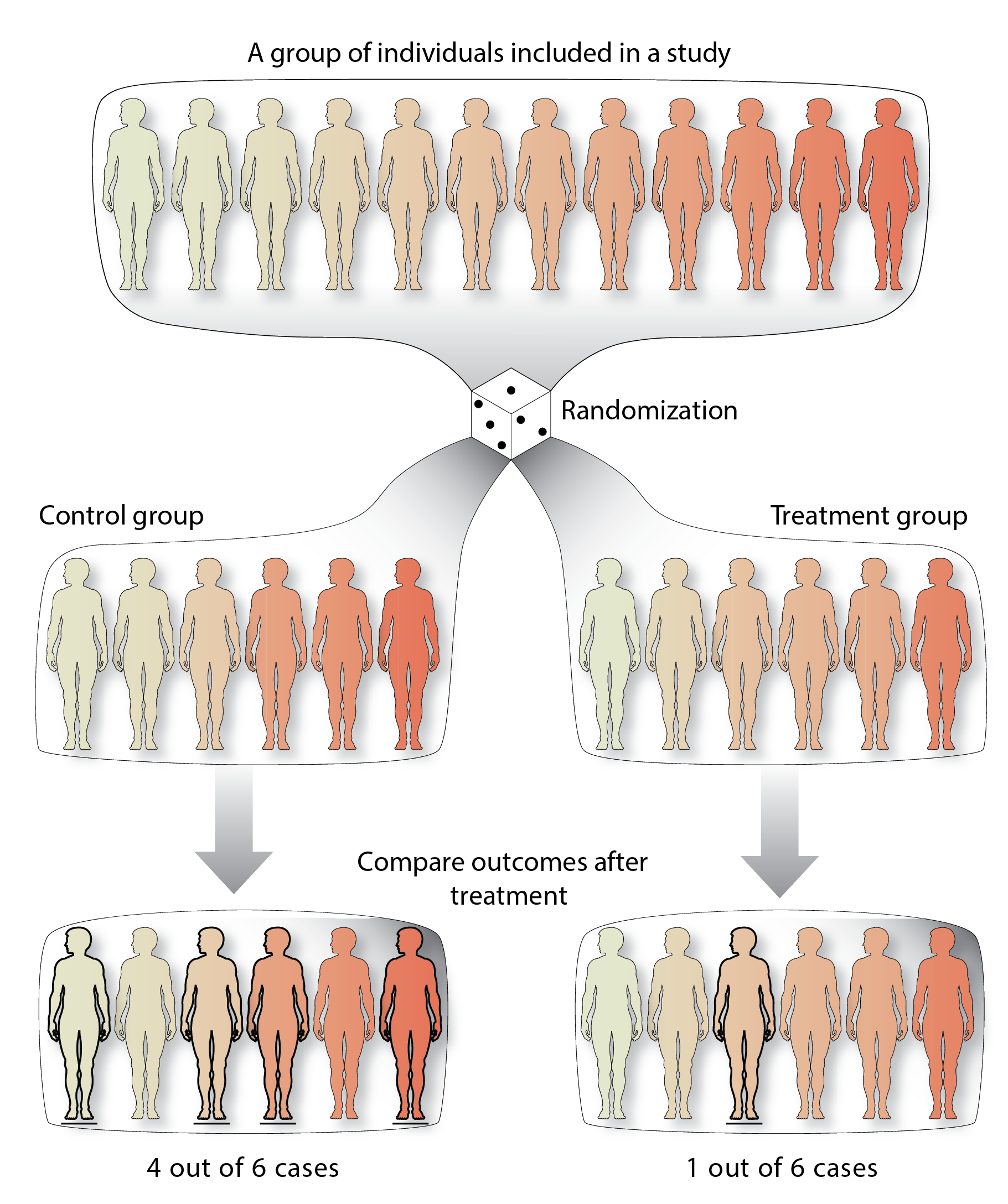
Figure 3: An example randomized control trial!
A group of individuals (top) is randomly assigned into two groups. The treatment group receives a treatment as an exposure (right), whereas the control group does not or receives a placebo treatment (left). At the end of the trial, the outcome is compared between the groups. In this case there is are different incidences of the disease between the two groups
It is infeasible to find causal relationships between all combinations of traits
Treatment-control experiments perturb potential risk factors, then assess if an outcome is also changed. We could ask ourselves, how many experiments would we need to carry out to fully understand the causal factors to human disease?
If we stand in the shoes of a pharmaceutical company, perhaps we would be interested in treating a disease with a drug. Not knowing where to start, we could do everything in a high-throughput fashion. Let’s design compounds for all the human protein-coding genes underlying all the GWAS loci that have been found for that disease and perform experiments to see if the drug would work. This would mean that the company has to design a compound that targets each of the 20,000 proteins in the human genome and subsequently run clinical trials for each of them to find all the drugs that may provide clinical benefit for a single disease (39). This is, of course, impossible in terms of money and time, as a single clinical trial usually costs a million US dollars and can run upwards of a billion US dollars [39,40].
Unfortunately, even considering all 20,000 protein coding genes (and thus 20,000 clinical trials) for a single disease would not be at all representative of all human molecular factors that may be important for disease. It would take 20,000 trials if we’re only interested in proteins and a single disease, but there are other biological factors that we may want to target with a drug. And if we’re interested in another disease, we would have to repeat all these trials again.
It is therefore crucial to find a way to prioritize which targets to perform experiments on, or alternatively, to identify candidate causal relationships from the results of observational and treatment‒control experiments
Epidemiological studies help us choose the right experimental targets
Of course, we already discussed how to pick the right target to perturb, we can use GWAS! Indeed, GWAS have been helpful in picking the right pharmaceutical drug targets [41].
Still, even with the help of GWAS results, identification of which target to perturb is non-trivial because we don’t know the underlying biological mechanism. Even today, very costly clinical trials fail due to efficacy. For example, the drug of interest can target the wrong biological factor, as has been shown in the failure of HDL-cholesterol treatments for heart disease.
One major observation in observational studies was that individuals who suffer from coronary artery disease have high LDL cholesterol levels, and low HDL cholesterol levels [8,42]. Initially, this prompted pharmaceutical companies to create medication to reduce LDL cholesterol levels in a patient. These trials were very successful in proving clinical benefit, and the resulting statin drugs are widely used to reduce the risk for heart disease [43].
The success of treating LDL cholesterol motivated pharmaceutical companies to create medication to increase HDL cholesterol levels, as this increase was also seen in similar epidemiological studies. Unfortunately drugs that targeted HDL cholesterol level increases failed to reduce the risk for coronary artery disease [44]. The trials that have shown the beneficial effect of LDL-cholesterol on heart disease were some of the largest successes in the pharmaceutical industry. While the trials testing the effect of HDL cholesterol were some of the most high profile failures of the pharmaceutical industry. Why were observational studies not able to differentiate between causal effects of LDL cholesterol and HDL cholesterol changes?
Observational studies are plagued by confounders, limiting their use in causal inference
If we zoom out a little, we can ask ourselves the question: Why can we find causal relationships using experiments and why can’t we do so using an observational study? After all, observational studies were the first to identify causal relationships like ‘smoking causes cancer’ and ‘LDL cholesterol causes heart disease’ [8].
The reason why observational studies are not always able to identify causal relationships, is because of what we call a confounder [45]. Confounders are factors that are usually unobserved that can cause a spurious correlation with your variables of interest(10) . It is important to note confounders are not necessarily limited to observational studies, but good experimental design can control for them during treatment-control studies(11).
Let’s consider a mock example with a confounder.If we plotted the amount of ice cream sales per day against the rate of drowning on any particular day, we could find a correlation that is reproducible across observational studies (Figure 4AB). Claiming a causal relationship from this evidence would result in the conclusion that ice cream causes drowning. Time to ban the sale of ice cream!
Of course ice cream consumption is not the cause of drowning (except for the rare ice cream‒induced stomach cramp tragedies). The cause of both ice cream sales and drowning is that the weather and location influences how many people are swimming and consuming a refreshing ice cream. In this mock example, warm sunny weather is the confounder. If we correct for the effect of weather when considering ice cream sales and drowning, the correlation would disappear (Figure 4C). In this mock example, we expect that weather may be a confounder, making it easy to control for its confounding effect when performing the analysis. Other variables that are common confounders are an individual’s sex or age and other intrinsic factors like body mass index (BMI), physical activity, education and smoking. Interestingly, even when the confounder is measured and known, causality could still be incorrectly claimed. Spurious correlation can occur through measurement error alone, indicating that claims to causality can still be incorrect even when all confounders are known and corrected for [46].
Unfortunately, very often we don’t know what all the confounders are in an analysis of causality. This is especially important when understanding human biology. As there are so many internal factors, many of them unmeasured and unstudied, it remains very difficult to anticipate all confounders.
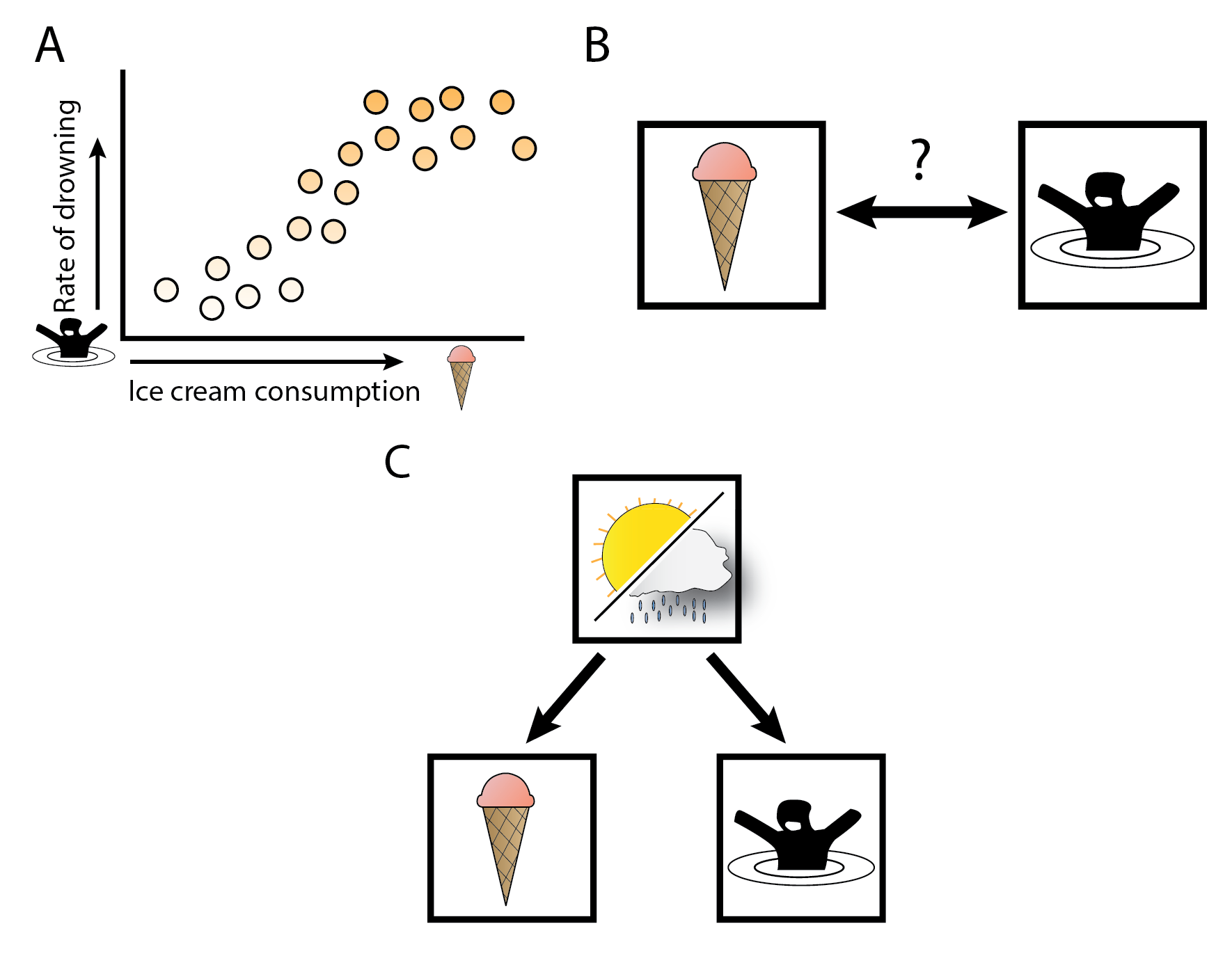
Figure 4: Correlations between ice cream consumption and drowning, and confounding.
(A) In this fictitious example, we show a correlation between ice cream consumption and rates of drowning. (B) This would indicate that there could be a causal relationship between ice-cream consumption and drowning. (C) However, there is no causal relationship between ice cream and drowning, but there is a single cause for both, namely how warm the weather is. The warm weather is the uncontrolled confounder in (A) and (B).
Treatment-control experiments remove confounding through randomization
Treatment‒control experiments are able to remove all confounders because of an important first step that happens before any observations can be made from the data. This is called a randomization step. An experiment on a group of individuals is first randomized into two or more groups that receive a different treatment. Only after this step is the outcome of interest observed. The randomization step ensures that any confounding that is present should be equally present between groups. We can then make causal conclusions about the data by comparing one group to another (Figure 3).
As an example, let’s construct an experiment where we put our mock hypothesis that ice cream consumption increases drowning to the test (12). We recruit a sufficiently large population close to a water source and divide it up into two groups. The treatment group receives an ice cream voucher from us, and the placebo group swears not to eat ice cream for the whole day. At the end of the day, we check back with each group and assess how many people have drowned. After the comparison, and ignoring the morbid nature of this outcome, we would not find a causal relationship because the consumption of ice cream does not cause individuals to drown. Note that we did not consider the confounder ‒ the weather ‒ in this experiment. The confounder has been accounted for through the randomization step, as the randomization makes ice cream consumption independent of the weather as a confounder.
Treatment‒control experiments are not limited to experiments on humans but are done daily in model systems and model organisms that compare one treatment to another. In all cases, the experimental setup removes confounding from the observations and will only evaluate the effect of the treatment of interest.
It’s important to note that the random nature of the experiment only needs to be random with respect to all the potential confounders, thereby removing the effects of the confounders themselves. But this does not mean that the randomization makes the groups fully random. Of course, it is not random which individuals receive the treatment.
Re-creating experimental designs in observational studies
Up to this point, I’ve discussed that experimental studies are expensive, a burden on the subjects and sometimes not feasible. Furthermore, observational studies do not find causality. So it seems as if we are stuck in an endless cycle of finding potential targets to perturb in observational studies and testing these targets in expensive experiments.
Fortunately, there is a way out of this vicious circle of observational association and expensive experimentation: we can mimic an experimental study in observational data so that causal relationships can be claimed.
Let’s again consider our mock example where ice cream is associated with drowning. We could use some random variables that affect ice cream consumption in the same way that we did in our experimental setup. We know that sugar tax rates influence how much ice cream is consumed. Looking across jurisdictions with and without sugar taxes, we can check if the introduction of the tax decreased ice cream consumption and then look at the rates of drowning across these jurisdictions. In this case, the sugar tax has had the same effect as the treatment in our hypothetical experiment: the tax influenced ice cream consumption, and we can look at the causal downstream effects. The tax increase is independent of our confounder, the weather, and so only looking at *tax-dependent ice cream consumption *would be independent of the weather, making it possible to find a causal relationship using this controlled ice cream consumption. This analysis would reveal that tax-dependent ice cream consumption is not a cause of drowning.
This is an example of a so-called *instrumental variable *(IV). IV analyses are widely used in econometrics to identify potential causal relationships. IV analysis tests for the causal effect of an *exposure *(ice cream consumption) towards an *outcome *(the rate of drowning). This allows for the identification of causal relationships that would not have been testable due to feasibility and cost. It is worth noting that IV analysis is a statistical technique that assumes certain conditions to be true. When these conditions are met, it can identify robust causal relationships. When these conditions are only partially (or not at all) satisfied, IV analysis finds, at best, nothing and, at worst, incorrect claims to causality.
Mendelian Randomization finds causal relationships in observational data
Mendelian randomization uses the randomness of genotypes to identify causality
So perhaps we can apply IV analysis to biological datasets to find causality? What sources of randomness are present in biological data that we can use? It may be easy to see that a human’s DNA is randomly allocated from their parents. There is no control on which parental alleles are passed to their progeny. Genetic variation is, in principle, random, and thus it could be used in IV analysis to identify causality. Moreover, DNA influences the majority of human traits [47–49] as well as molecular mechanisms, such as gene expression or protein levels [35,50–52]. So if we have an observational cohort with DNA marker data information available, we could use it’s randomness to identify causal relationships from observational data. This method is called *Mendelian randomization *(MR), named after Gregor Mendel, who first observed genetic inheritance patterns [53,54].
To give an example on how this would work, let’s construct a relatively simple MR experiment where we’re interested in the causal effect of the LDL-cholesterol biomarker as an *exposure (or risk factor) *on the incidence of heart disease as the outcome (the result of interest).
We take an observational study where we know the LDL cholesterol concentrations of individuals and if they have had heart disease. First, we determine which genetic variants are associated with variations in LDL cholesterol levels. Detection of these variants is done using GWAS. After we select a variant to use as an IV for our analysis, we separate the whole cohort into three groups of people depending on their genotype for the variant (i.e. homozygous for the reference allele, heterozygous and homozygous for the alternative allele). Then, we see if there are differences in heart disease incidence between each group. If the experiment was performed correctly, a significant difference in heart disease incidence between each group would indicate that there is a causal relationship (Figure 5A).
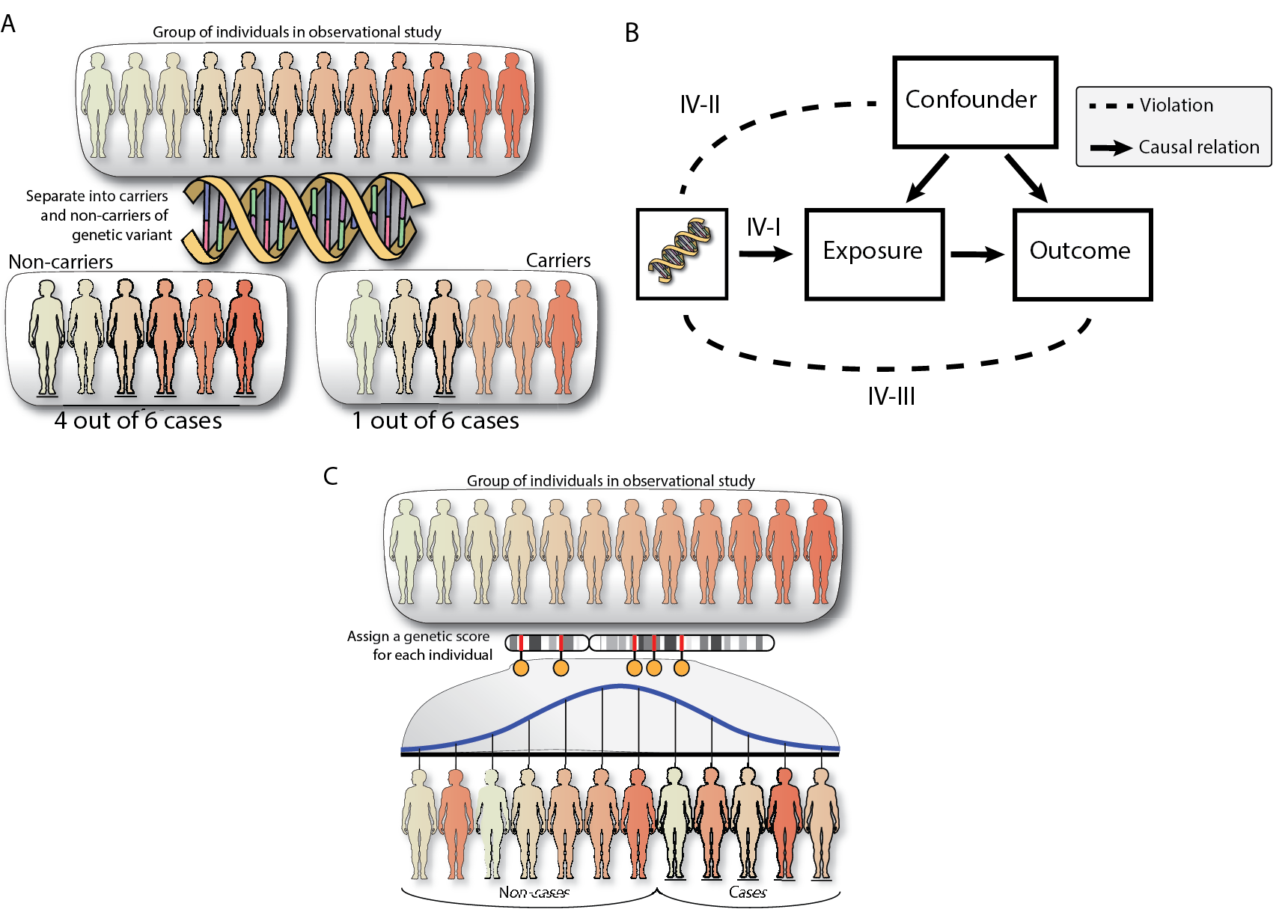
Figure 5: Mendelian Randomization explanation and schema with assumptions
(A) Mendelian randomization can be used in observational cohorts with DNA information available. In the simplest example, Mendelian randomization would separate individuals into carriers and non-carriers to identify how many cases of a specific disease are in both groups. (B) For more power, we can provide a risk score to each individual based on multiple genetic variants and then perform MR. (C) Mendelian randomization will only identify causality when none of the assumptions are violated: IV-I requires that the genetic variant chosen as IV has an effect on the exposure (Strong instruments), IV-II requires that the genetic variants are not influenced by the confounder (Independence) and IV-III requires that the genetic variant only has an effect on the outcome through the exposure (exclusion restriction).
When is Mendelian randomization appropriate for causal inference?
If we want to rely on the inferences of MR, it is important to understand how and when MR fails. As with any statistical technique, the underlying theory behind MR (IV analysis) requires that three assumptions are met (Figure 5C):
- IV-I: The instrumental variable is robustly associated to the exposure variable (Strong instrument)
- IV-II: The instrumental variable is not influenced by any confounder (Independence)
- IV-III: The instrumental variable is only associated to the outcome through the exposure (Exclusion)
I will approach these assumptions from a treatment-control experiment perspective and discuss how these assumptions may fail in the human genetics perspective to help understand cases when these assumptions fail, and how often this happens when genetic variants are used as IVs.
The IV-I assumption is violated when an IV does not have a sufficiently large effect on the exposure
The IV-I assumption requires that the instrumental variable is actually associated with changes in the exposure. If we consider the treatment-control experiment, we shouldn’t be providing the treatment group too low a dose of our new treatment, i.e. there should be a difference between the treatment and control groups.
In experimental designs, it is usually known what kind of effect the treatment has as these have been at least broadly tested in animal studies. This is not always the case in MR. The genetic variants we use are statistically associated with an exposure trait. Sometimes, these associations are not robust, or are even false positives, and using them as an instrument will result in invalid causal inference. When a genetic variant is selected as an instrument, MR will not question it’s validity. There are ways to reduce the bias from these weak instruments and these sources of bias can be eliminated by being very stringent about what variants to select for MR.
We can, for example, i) use very stringent cutoffs of association significance to select only robustly associated variants, ii) require that these associations were replicated in an independent cohort, and iii) potentially perform the selection of IV in a cohort independent from the one that will be used for MR, as confounding correlations with the outcome will be absent [55].
The IV-II assumption is violated through population stratification and selection in MR
The IV-II assumption requires that an IV is independent of any confounders. In other words the IV should be randomized with respect to any confounders. In the same way that we shouldn’t put all the smokers in the treatment group in a treatment-control experiment, the IV-II assumption requires that we are independent from any potential confounders (Figure 5B).
In experimental designs it’s fairly obvious that incorrect randomization would invalidate an experiment, but when and how do we violate the IV-I assumption when we use genetic variation as our IV? In general, the random segregation of alleles is ensured by evolutionary forces, and we assume this happens in human genetics [56]. But we could think of selection processes that may influence the viability of individuals with certain variants. We know selection happens in the human genome [57,58]. Fortunately selection effects are usually tested for in the genome, using the Hardy-Weinberg equilibrium test [59]. This makes it unlikely that selection or population stratification has large effects that will violate the assumptions of MR.
Next to selection processes, there is a chance that IV-II could be violated through a process called collider bias [60,61]. Very often, we determine which variant to use in MR through a GWAS study. These GWAS studies have pre-processed their phenotypes. If the phenotype was adjusted for a confounder in the analysis, this could make the resulting IV association also dependent on the confounder which violates IV-II. If, however, the phenotype was not corrected for covariates, collider bias is not an issue.
The IV-III assumption is violated when an IV affects multiple traits
that are causal to the outcome.
The IV-III assumption states that there should only be a single path from the IV to the exposure to the outcome (Figure 5B) a condition also known as the exclusion restriction and lack of (horizontal) pleiotropy assumption(13). If there is any other path from the SNP to the outcome that does not pass through the exposure but rather through another phenotype, the IV-III assumption is violated.
In a treatment‒control experiment, IV-III could be violated if the drug treatment has some sort of contamination in it in addition to the compound of interest. Thus, strictly speaking, any effect that is observed in the experiment cannot be purely attributable to the compound of interest since it could also be due to the contamination that is part of the treatment.
The IV-III assumption is perhaps the most difficult to account for when using genetic instruments. We don’t control which variant is a potential IV for our exposure, and it could be that the IV also has an effect on hundreds of other traits, which all could violate IV-III.
Ensuring that there are no known GWAS signals on your IV of interest is important but not sufficient to control for IV-III because there could be other unobserved factors that are regulated by our genetic variant used as an IV. IV-III cannot be ignored in an MR study, and it represents one of the major hurdles of this approach. Luckily, there are ways to reduce the effects of the IV-III when we apply MR on multiple variants.
Mendelian randomization is fairly easy to apply to genetic epidemiological studies.
Usually the most difficult part of an IV analysis is identifying informative IVs. This is not necessarily the case for an MR analysis. Thanks to large-scale genomic studies, many genetic variants have been found that can be used for MR, across many different phenotypes. MR can use multiple GWAS-associated variants together to improve the causal estimate (Figure 5C). Meta-analysis techniques coupled with sensitivity analyses can be used to identify average causal effects across multiple IVs, to investigate if IV-III was violated by some of them and eventually to recalculate the causal relationship without these pleiotropic IVs [54,60,62–65].
Furthermore, MR can work in a ‘two-sample’ fashion, meaning that the exposure and outcome can be estimated in different datasets. This allows for causal inference across two independent observational studies. Moreover, it is not even necessary to perform MR on individual level data, as GWAS summary statistics can be used, allowing for causal inference without having access to individual-level data. Web tools that compile summary statistics and run MR are widely available [66,67].
Mendelian randomization has identified and disproved causal relationships
Easy applicability and the attractiveness of being able to claim causality has made MR a popular method for researchers in human biology and in the social sciences. Indeed MR has been successfully applied to identify causal relationships in humans without having to perform costly experiments. MR confirmed the well known causal relationship of LDL-cholesterol causes heart disease, and it was one of the first methods that has disproved the causal relationship between HDL-cholesterol and heart disease [44]. Another relationship that MR has disproved is the causal influence of vitamin D intake and the incidence of cancer [68,69].
These results showcase how MR has been able to identify causal relationships between easily observable complex traits. But, perhaps the main promise of the method is that there is no limitation to which traits can be assessed. MR is possible as long as there are variants that are suitable IVs. As sample sizes increase in large cohort‒based GWAS and more traits are investigated, we are finding more and more genetic loci associated with all sorts of traits, with robust effects (to satisfy IV-I). The availability of more genetic variants to use together as IVs in a combined meta-analysis approach also make MR analyses more and more powerful.
Mendelian randomization on -omics phenotypes.
I find there is particular promise for MR in molecular phenotypes. We know that -omics phenotypes are difficult to interpret through association studies. Furthermore, treatment-control experiments that test the influence of one molecular factor on all other molecular factors are expensive and time-consuming. MR has the potential to identify the same causal effects as these experiments and is therefore in a great position to understand how -omics phenotypes are related to one another *as well as how -omics traits are related *to complex human traits such as disease.
However MR on -omics traits is not as straightforward as MR on complex traits. For instance, the number of available IVs is more limited in -omics analysis, making it difficult to know if the IV follows all the assumptions underlying MR. There is also the issue of extensive quality control that needs to occur in -omics traits, which may be a cause of collider bias in MR.
That is not to say that it is impossible to perform MR on -omics molecular phenotypes and the potential payoff for finding robust causality is very large. For example, we have benefited tremendously from the knowledge of the metabolic pathways such as the Krebbs cycle [70]. Having similar network structures for -omics data layers could help us in understanding the human biological system even more.
References
[1. Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walter P. Molecular Biology of the Cell. Taylor & Francis Group; 2007. 1732 p.]
[2. Aghababian. Essentials of Emergency Medicine. Jones & Bartlett Publishers; 2010. 1092 p.]
[3. Murray CJL, Abbafati C, Abbas KM, Abbasi M, Abbasi-Kangevari M, Abd-Allah F, et al. Five insights from the Global Burden of Disease Study 2019. The Lancet. 2020 Oct 17;396(10258):1135–59.]
[4. CDC. Different COVID-19 Vaccines [Internet]. Centers for Disease Control and Prevention. 2021 [cited 2021 Apr 22]. Available from: https://www.cdc.gov/coronavirus/2019-ncov/vaccines/different-vaccines.html]
[5. GLANVILLE D. COVID-19 vaccines: authorised [Internet]. European Medicines Agency. 2021 [cited 2021 Apr 22]. Available from: https://www.ema.europa.eu/en/human-regulatory/overview/public-health-threats/coronavirus-disease-covid-19/treatments-vaccines/vaccines-covid-19/covid-19-vaccines-authorised]
[6. Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell. 2020 Apr 16;181(2):271-280.e8.]
[7. Dai L, Gao GF. Viral targets for vaccines against COVID-19. Nat Rev Immunol. 2021 Feb;21(2):73–82.]
[8. Mahmood SS, Levy D, Vasan RS, Wang TJ. The Framingham Heart Study and the epidemiology of cardiovascular disease: a historical perspective. The Lancet. 2014 Mar 15;383(9921):999–1008.]
[9. Sudlow C, Gallacher J, Allen N, Beral V, Burton P, Danesh J, et al. UK Biobank: An Open Access Resource for Identifying the Causes of a Wide Range of Complex Diseases of Middle and Old Age. PLOS Med. 2015 Mar 31;12(3):e1001779.]
[10. Scholtens S, Smidt N, Swertz MA, Bakker SJL, Dotinga A, Vonk JM, et al. Cohort Profile: LifeLines, a three-generation cohort study and biobank. Int J Epidemiol. 2015 Aug;44(4):1172–80.]
[11. Tigchelaar EF, Zhernakova A, Dekens JAM, Hermes G, Baranska A, Mujagic Z, et al. Cohort profile: LifeLines DEEP, a prospective, general population cohort study in the northern Netherlands: study design and baseline characteristics. BMJ Open. 2015 Aug 1;5(8):e006772.]
[12. Cederholm T, Palmblad J. Are omega-3 fatty acids options for prevention and treatment of cognitive decline and dementia? Curr Opin Clin Nutr Metab Care. 2010 Mar;13(2):150–5.]
[13. Sala-Vila A, Calder PC. Update on the Relationship of Fish Intake with Prostate, Breast, and Colorectal Cancers. Crit Rev Food Sci Nutr. 2011 Oct 1;51(9):855–71.]
[14. Suratannon N, van Wijck RTA, Broer L, Xue L, van Meurs JBJ, Barendregt BH, et al. Rapid Low-Cost Microarray-Based Genotyping for Genetic Screening in Primary Immunodeficiency. Front Immunol [Internet]. 2020 [cited 2021 May 3];11. Available from: https://www.frontiersin.org/articles/10.3389/fimmu.2020.00614/full]
[15. Lappalainen T, Sammeth M, Friedländer MR, ‘t Hoen PAC, Monlong J, Rivas MA, et al. Transcriptome and genome sequencing uncovers functional variation in humans. Nature. 2013 Sep;501(7468):506–11.]
[16. Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods. 2013 Dec;10(12):1213–8.]
[17. Auton A, Abecasis GR, Altshuler DM, Durbin RM, Abecasis GR, Bentley DR, et al. A global reference for human genetic variation. Nature. 2015 Oct;526(7571):68–74.]
[18. Bonder MJ, Smail C, Gloudemans MJ, Frésard L, Jakubosky D, D’Antonio M, et al. Identification of rare and common regulatory variants in pluripotent cells using population-scale transcriptomics. Nat Genet. 2021 Mar;53(3):313–21.]
[19. van der Wijst MGP, Brugge H, de Vries DH, Deelen P, Swertz MA, Franke L. Single-cell RNA sequencing identifies celltype-specific cis-eQTLs and co-expression QTLs. Nat Genet. 2018 Apr;50(4):493–7.]
[20. Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010 Mar;464(7285):59–65.]
[21. Hasin Y, Seldin M, Lusis A. Multi-omics approaches to disease. Genome Biol. 2017 May 5;18(1):83.]
[22. Bonder MJ, Kurilshikov A, Tigchelaar EF, Mujagic Z, Imhann F, Vila AV, et al. The effect of host genetics on the gut microbiome. Nat Genet. 2016 Nov;48(11):1407–12.]
[23. Gallagher MD, Chen-Plotkin AS. The Post-GWAS Era: From Association to Function. Am J Hum Genet. 2018 May 3;102(5):717–30.]
[24. Buniello A, MacArthur JAL, Cerezo M, Harris LW, Hayhurst J, Malangone C, et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 2019 Jan 8;47(D1):D1005–12.]
[25. Schaub MA, Boyle AP, Kundaje A, Batzoglou S, Snyder M. Linking disease associations with regulatory information in the human genome. Genome Res. 2012 Sep 1;22(9):1748–59.]
[26. Maurano MT, Humbert R, Rynes E, Thurman RE, Haugen E, Wang H, et al. Systematic Localization of Common Disease-Associated Variation in Regulatory DNA. Science. 2012 Sep 7;337(6099):1190–5.]
[27. Leeuw CA de, Mooij JM, Heskes T, Posthuma D. MAGMA: Generalized Gene-Set Analysis of GWAS Data. PLOS Comput Biol. 2015 Apr 17;11(4):e1004219.]
[28. Pers TH, Karjalainen JM, Chan Y, Westra H-J, Wood AR, Yang J, et al. Biological interpretation of genome-wide association studies using predicted gene functions. Nat Commun. 2015 Jan 19;6(1):5890.]
[29. Locke AE, Kahali B, Berndt SI, Justice AE, Pers TH, Day FR, et al. Genetic studies of body mass index yield new insights for obesity biology. Nature. 2015 Feb 12;518(7538):197–206.]
[30. Wahbeh H, Radin D, Yount G, Woodley of Menie MA, Sarraf MA, Karpuj MV. Genetics of psychic ability - A pilot case-control exome sequencing study. EXPLORE [Internet]. 2021 Mar 1 [cited 2021 May 6]; Available from: https://www.sciencedirect.com/science/article/pii/S1550830721000501]
[31. Giambartolomei C, Vukcevic D, Schadt EE, Franke L, Hingorani AD, Wallace C, et al. Bayesian Test for Colocalisation between Pairs of Genetic Association Studies Using Summary Statistics. PLOS Genet. 2014 May 15;10(5):e1004383.]
[32. Hormozdiari F, van de Bunt M, Segrè AV, Li X, Joo JWJ, Bilow M, et al. Colocalization of GWAS and eQTL Signals Detects Target Genes. Am J Hum Genet. 2016 Dec 1;99(6):1245–60.]
[33. Ongen H, Brown AA, Delaneau O, Panousis NI, Nica AC, Dermitzakis ET. Estimating the causal tissues for complex traits and diseases. Nat Genet. 2017 Dec;49(12):1676–83.]
[34. Morris JA, Daniloski Z, Domingo J, Barry T, Ziosi M, Glinos DA, et al. Discovery of target genes and pathways of blood trait loci using pooled CRISPR screens and single cell RNA sequencing. bioRxiv. 2021 Apr 8;2021.04.07.438882.]
[35. Võsa U, Claringbould A, Westra H-J, Bonder MJ, Deelen P, Zeng B, et al. Unraveling the polygenic architecture of complex traits using blood eQTL metaanalysis. bioRxiv. 2018 Oct 19;447367.]
[36. Montgomery DC. Design and Analysis of Experiments. John Wiley & Sons Incorporated; 2013. book.]
[37. Greenland S. Randomization, Statistics, and Causal Inference. Epidemiology. 1990;1(6):421–9.]
[38. Riegelman R. Contributory cause: Unnecessary and insufficient. Postgrad Med. 1979 Aug 1;66(2):177–9.]
[39. Sertkaya A, Wong H-H, Jessup A, Beleche T. Key cost drivers of pharmaceutical clinical trials in the United States. Clin Trials. 2016 Apr 1;13(2):117–26.]
[40. DiMasi JA, Hansen RW, Grabowski HG. The price of innovation: new estimates of drug development costs. J Health Econ. 2003 Mar 1;22(2):151–85.]
[41. Plenge RM, Scolnick EM, Altshuler D. Validating therapeutic targets through human genetics. Nat Rev Drug Discov. 2013 Aug;12(8):581–94.]
[42. Lloyd-Jones DM, O’Donnell CJ, D’Agostino RB, Massaro J, Silbershatz H, Wilson PWF. Applicability of Cholesterol-Lowering Primary Prevention Trials to a General Population: The Framingham Heart Study. Arch Intern Med. 2001 Apr 9;161(7):949.]
[43. Adhyaru BB, Jacobson TA. Safety and efficacy of statin therapy. Nat Rev Cardiol. 2018 Dec;15(12):757–69.]
[44. Rader DJ, Hovingh GK. HDL and cardiovascular disease. The Lancet. 2014 Aug 16;384(9943):618–25.]
[45. Greenland S, Pearl J, Robins JM. Confounding and Collapsibility in Causal Inference. Stat Sci. 1999 Feb;14(1):29–46.]
[46. Fewell Z, Davey Smith G, Sterne JAC. The Impact of Residual and Unmeasured Confounding in Epidemiologic Studies: A Simulation Study. Am J Epidemiol. 2007 Sep 15;166(6):646–55.]
[47. Shin S-Y, Fauman EB, Petersen A-K, Krumsiek J, Santos R, Huang J, et al. An atlas of genetic influences on human blood metabolites. Nat Genet. 2014 Jun;46(6):543–50.]
[48. Ge T, Chen C-Y, Neale BM, Sabuncu MR, Smoller JW. Phenome-wide heritability analysis of the UK Biobank. PLOS Genet. 2017 Apr 7;13(4):e1006711.]
[49. Tegegne Balewgizie S., Man Tengfei, van Roon Arie M., Asefa Nigus G., Riese Harriëtte, Nolte Ilja, et al. Heritability and the Genetic Correlation of Heart Rate Variability and Blood Pressure in >29 000 Families. Hypertension. 2020 Oct 1;76(4):1256–62.]
[50. Sinnott-Armstrong N, Tanigawa Y, Amar D, Mars N, Benner C, Aguirre M, et al. Genetics of 35 blood and urine biomarkers in the UK Biobank. Nat Genet. 2021 Feb;53(2):185–94.]
[51. Sun BB, Maranville JC, Peters JE, Stacey D, Staley JR, Blackshaw J, et al. Genomic atlas of the human plasma proteome. Nature. 2018 Jun;558(7708):73–9.]
[52. Consortium TGte. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science. 2020 Sep 11;369(6509):1318–30.]
[53. Katan M. APOUPOPROTEIN E ISOFORMS, SERUM CHOLESTEROL, AND CANCER. The Lancet. 1986 Mar 1;327(8479):507–8.]
[54. Smith GD. Mendelian Randomization for Strengthening Causal Inference in Observational Studies: Application to Gene × Environment Interactions. Perspect Psychol Sci. 2010 Sep 1;5(5):527–45.]
[55. Burgess S, Thompson SG, CRP CHD Genetics Collaboration. Avoiding bias from weak instruments in Mendelian randomization studies. Int J Epidemiol. 2011 Jun;40(3):755–64.]
[56. Edwards AWF. Natural Selection and the Sex Ratio: Fisher’s Sources. Am Nat. 1998 Jun 1;151(6):564–9.]
[57. Bamshad M, Wooding SP. Signatures of natural selection in the human genome. Nat Rev Genet. 2003 Feb;4(2):99–110.]
[58. Beauchamp JP. Genetic evidence for natural selection in humans in the contemporary United States. Proc Natl Acad Sci. 2016 Jul 12;113(28):7774–9.]
[59. Weir BS. Genetic data analysis. Methods for discrete population genetic data. Genet Data Anal Methods Discrete Popul Genet Data [Internet]. 1990 [cited 2021 May 18]; Available from: https://www.cabdirect.org/cabdirect/abstract/19900180990]
[60. Burgess S, Thompson SG. Mendelian Randomization: Methods for Using Genetic Variants in Causal Estimation. Taylor & Francis; 2015. 224 p.]
[61. Barry C, Liu J, Richmond R, Rutter MK, Lawlor DA, Dudbridge F, et al. Exploiting collider bias to apply two-sample summary data Mendelian randomization methods to one-sample individual level data. medRxiv. 2020 Oct 23;2020.10.20.20216358.]
[62. Bowden J, Davey Smith G, Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol. 2015 Apr 1;44(2):512–25.]
[63. Verbanck M, Chen C-Y, Neale B, Do R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat Genet. 2018 May;50(5):693.]
[64. Burgess S, Thompson SG. Multivariable Mendelian Randomization: The Use of Pleiotropic Genetic Variants to Estimate Causal Effects. Am J Epidemiol. 2015 Feb 15;181(4):251–60.]
[65. Rees JMB, Wood AM, Burgess S. Extending the MR-Egger method for multivariable Mendelian randomization to correct for both measured and unmeasured pleiotropy. Stat Med. 36(29):4705–18.]
[66. Hemani G, Zheng J, Elsworth B, Wade KH, Haberland V, Baird D, et al. The MR-Base platform supports systematic causal inference across the human phenome. Loos R, editor. eLife. 2018 May 30;7:e34408.]
[67. Kamat MA, Blackshaw JA, Young R, Surendran P, Burgess S, Danesh J, et al. PhenoScanner V2: an expanded tool for searching human genotype-phenotype associations. Bioinforma Oxf Engl. 2019 Nov 1;35(22):4851–3.]
[68. Ong J-S, Gharahkhani P, An J, Law MH, Whiteman DC, Neale RE, et al. Vitamin D and overall cancer risk and cancer mortality: a Mendelian randomization study. Hum Mol Genet. 2018 Dec 15;27(24):4315–22.]
[69. Chandler PD, Chen WY, Ajala ON, Hazra A, Cook N, Bubes V, et al. Effect of Vitamin D ][ Supplements on Development of Advanced Cancer: A Secondary Analysis of the VITAL Randomized Clinical Trial. JAMA Netw Open. 2020 Nov 18;3(11):e2025850.]
[70. Krebs HA, Johnson WA. Metabolism of ketonic acids in animal tissues. Biochem J. 1937 Apr 1;31(4):645–60.]
Footnotes
(1): Although this knowledge would be needed to identify those individuals who will not benefit from paracetamol or who will develop side effects.
(2): Genetic variation describes all the places on the DNA with individual variation.
(3): Gene expression is the measurement of how much a gene is transcribed from the DNA: How much is a specific gene ‘turned on’ or ‘turned off’
(4): -omics is a way to represent branches of biological study that focus on a specific molecular mark.
(5): Multi-omics is the study of multiple layers of -omics data together
(6): Sometimes also known as gluten allergy. Individuals with celiac disease experience a strong immune response in the small intestine upon ingestion of gluten. Celiac disease is an autoimmune disease.
(7): A protein isoform is a specific version of a protein that is the result of a genetic difference.
(8): Heritability means that a certain phenotype runs in families. A more statistically sound explanation would be the amount of variance of a phenotype that can be explained by an individual’s DNA.
(9): Which is really the same as filling out that you’re psychic on a questionnaire.
(10): Confounders are sometimes also called ‘confusors’, as they can show a causal relationship that is not there
(11): Confounding can also occur in treatment control experiments if the groups are imbalanced with respect to the confounder. For instance if the control group contains more smokers.
(12): This is an example of a negative result. In finding causal relationships in biology, negative results are very important as the risk of false positives is so high. Therefore it’s important to understand how negative results come about
(13): In this thesis, we mostly refer to a violation of IV-III as the presence of pleiotropy.
Last modified on 2022-09-19